What Is Epigenetics?
An overview of epigenetic regulations and analysis tools
What Is Epigenetics?
Epigenetics is the study of changes produced in gene expression caused by mechanisms other than changes in the underlying DNA sequence. But what exactly does this mean?
All multicellular organisms (like you!) contain roughly the same DNA sequence within nuclei across all cell types, but skin cells can be uniquely distinguished from liver hepatocytes and heart cardiomyocytes for example. The mechanisms behind the establishment of these specific cell types were first recognized as “epigenetic” regulation.
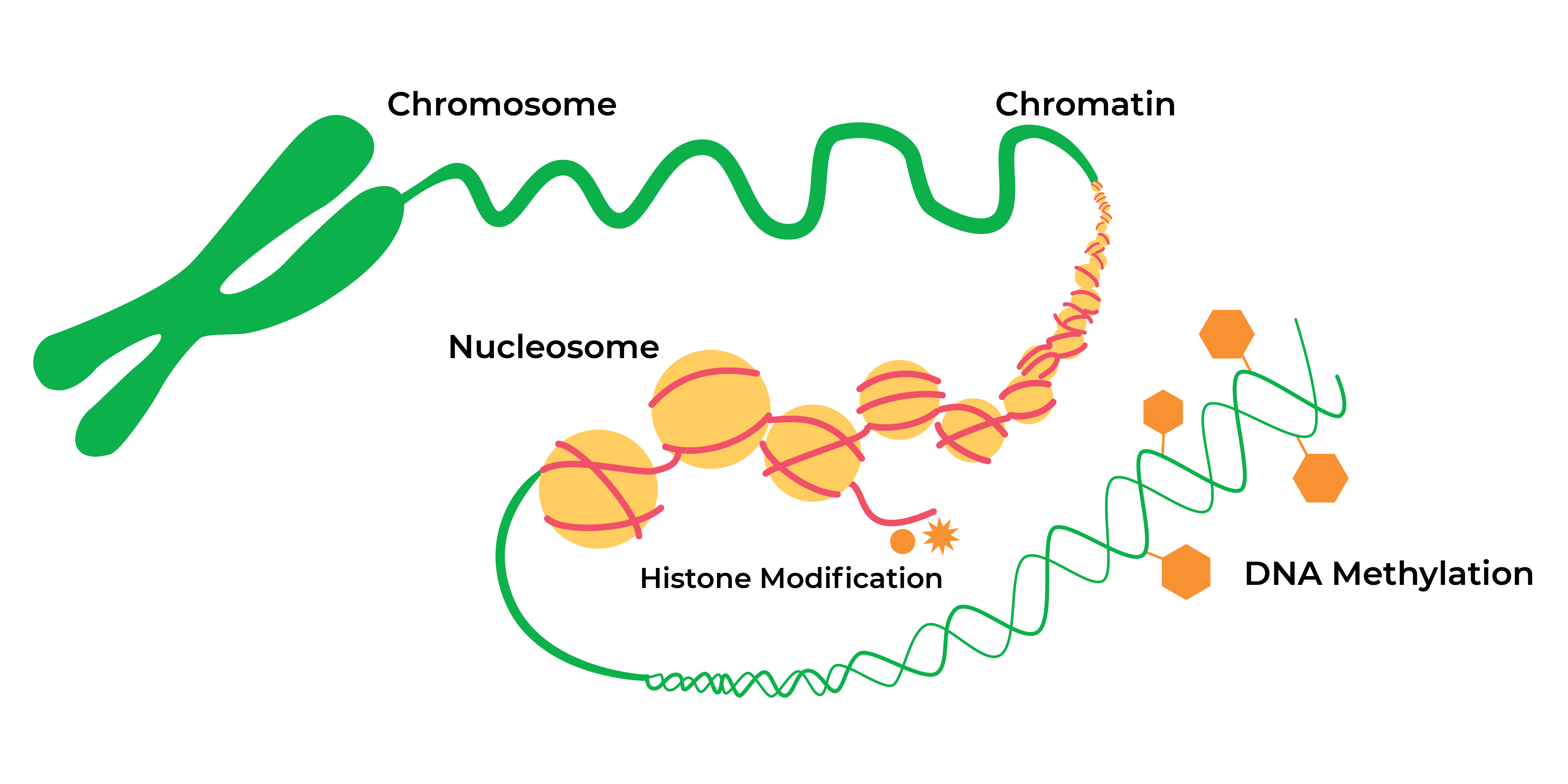
The Greek prefix “epi-” means “on” or “over” the DNA sequence itself. After decades of research into these epigenetic modifications or “marks”, nowadays epigenetics in general refers to heritable histone modifications and DNA methylation without changes the underlying DNA sequence which can affect gene activity.
The landscape of epigenetic regulation has expanded vastly. Histone modifications (such as acetylation and methylation of lysine residues within core proteins) serve to expand or contract the chromatin into “open” or “closed” configurations associated with “on” or “off” gene expression. Expression of certain non-coding RNAs serve to help regulate gene expression during or after transcription. DNA methylation is another key modification whereby methyl groups are added to cytosines positioned within cytosine-phosphate-guanine (CpG) sequences across the genome.
What is the function of epigenetic modifications?

The ultimate function of all these different marks is to regulate gene expression in a coordinated fashion. Research now indicates these marks serve as a molecular mechanism that mediates the influence of the environment on the genome beginning from embryonic development. For example, the lifestyle and environmental conditions of the mother during early pregnancy may have long-term effects on the health of the offspring.1 The DNA methylome of newborns has been reported to be affected by maternal smoking.2 In fact, there is emerging evidence that some epigenetic marks are heritable across generations, even though much of these marks are first erased in the very early embryo. At birth and as we age, DNA methylation patterns continue to change over time. Recent research has also found that methylation levels at specific loci can be used as a predictor of a person’s biological age.3
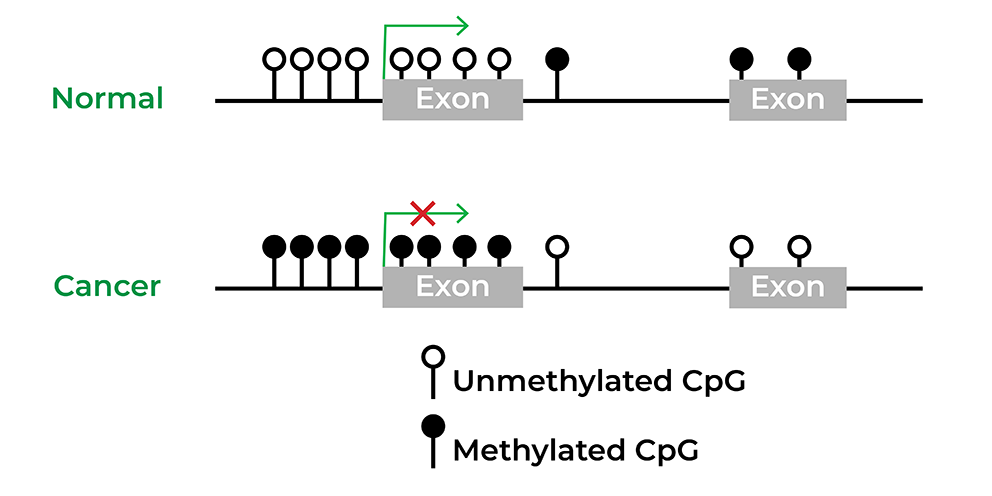
Epigenetics has also been widely implicated in diseases, particularly in cancer. Several initiatives, such as The Cancer Genome Atlas or TCGA, were rapidly developed to obtain the epigenomes of human disorders. Utilizing these large cohort studies, biomarkers have been discovered and even commercialized for various cancers from colon cancer to bladder cancer.4 Epigenetic drugs have proven to be effective for the treatment of hematological and solid tumors, and there are many clinical trials ongoing to develop small-molecule inhibitors that target the cancer epigenome alone or combined with other therapy.5
How do you study epigenetic changes?
The most well-characterized epigenetic mark is DNA methylation. Standard sequencing methods do not work for analyzing DNA methylation directly because the methyl group is covalently bound to the cytosine (5-methylcytosine; 5-mC). Bisulfite conversion is considered the gold standard for identifying changes in 5-mC. This chemical process deaminates cytosines and converts them to uracil unless methyl groups are present on the cytosine base. Using Next-Gen Sequencing (NGS), the methylated cytosines are read as “C” in the sequence, while cytosines that are converted to uracil are read as “T”. By comparing these sequences to a reference genome, one can calculate the percentage of methylation at a single-base resolution. Reduced Representation Bisulfite Sequencing (RRBS), Whole-Genome Bisulfite Sequencing (WGBS) and targeted bisulfite sequencing are the most wildly used NGS technologies to profile the methylome.
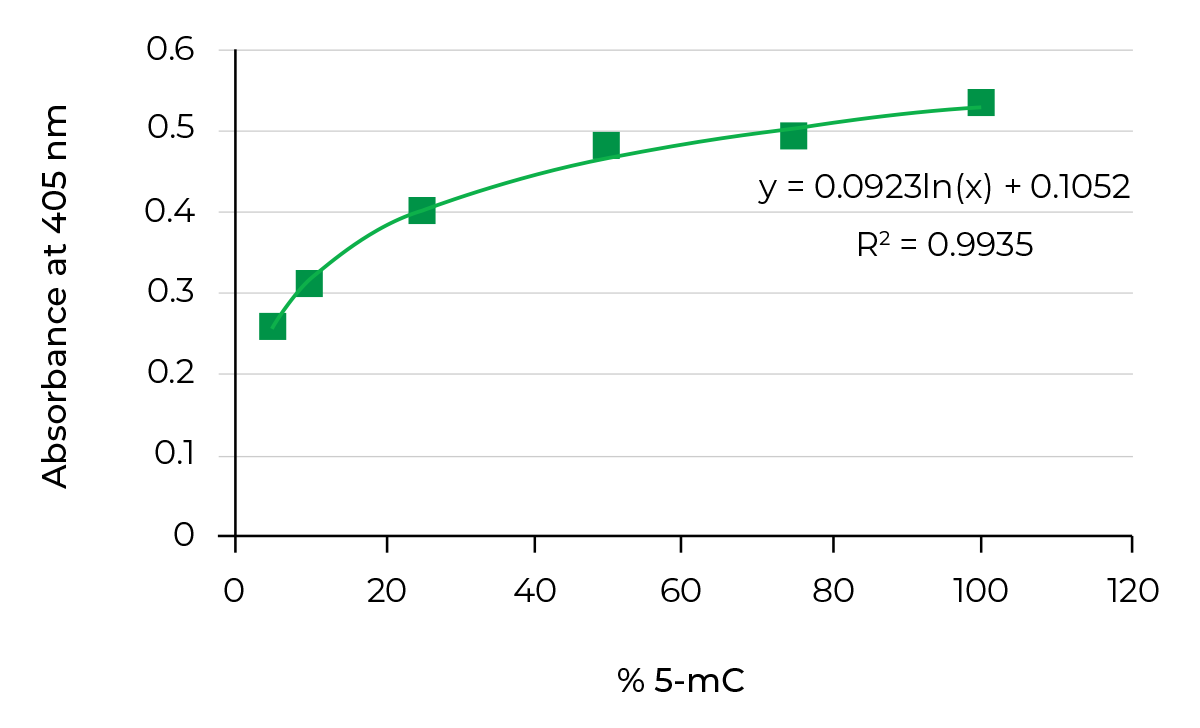
On the other hand, bisulfite-free methods, such as Methylated DNA immunoprecipitation(MeDIP), utilize antibodies targeting 5-mC to enrich methylated DNA from a pool of fragmented genomic DNA. This enriched fraction can then be used in genome-wide methylation analysis. Researchers could also quantify 5-methylcytosine (5-mC) DNA from a variety of samples in a high-throughput manner by using the same antibodies to target 5-mC combined with ELISA technology.
How to Analyze Chromatin Structure Changes
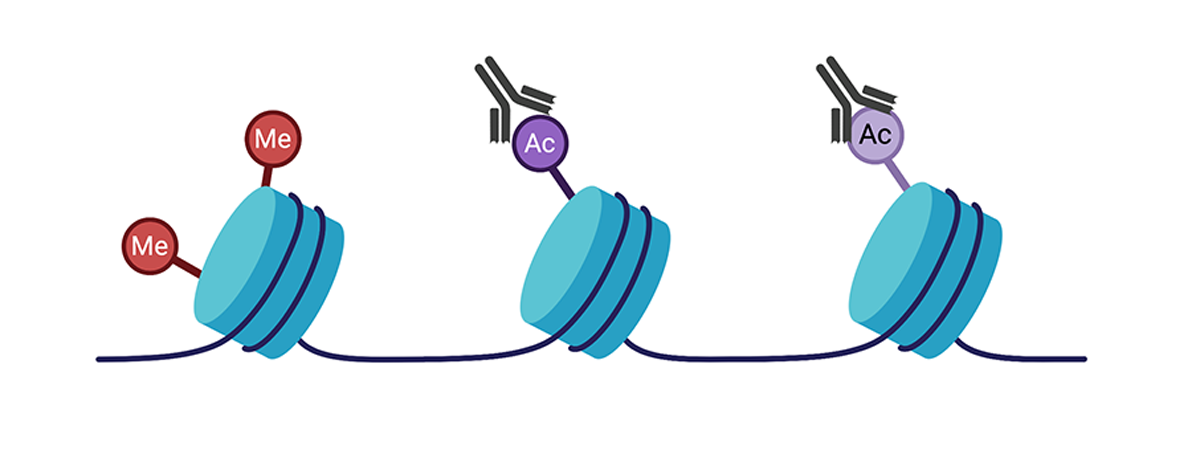
Histone modifications are also covalent modifications made to the core histone proteins, which may alter gene expression as well. Following crosslinking of proteins to DNA, protein-specific antibodies can be used to selectively precipitate DNA fragments that are bound to the protein in a process known as chromatin immunoprecipitation (or ChIP). The enriched DNA fragments can then be sequenced (ChIP-seq) by Next-Gen Sequencing to achieve a genome wide profile of the specific histone modification. ChIP-seq methods can also be used to analyze transcription factors and other enzyme binding sites in the genome.
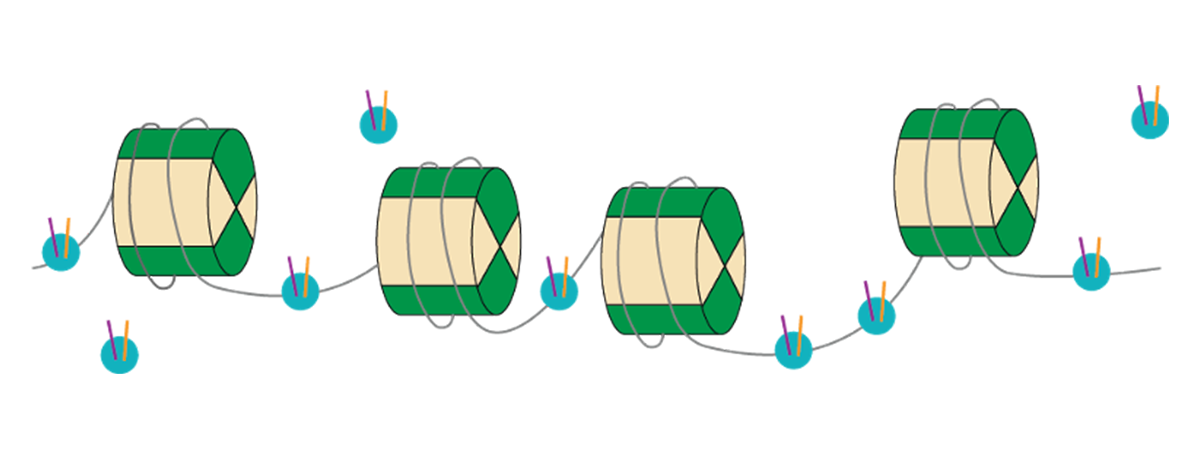
As chromatin accessibility in its “open” configuration is typically associated with gene expression, methods that measure the presence or absence of open chromatin help us to understand the importance of chromatin structure changes of the epigenetic landscape. The most commonly used technique for chromatin accessibility is ATAC-seq (or Assay for Transposase-Accessible Chromatin), which allows researchers to look at the accessibility of DNA. This powerful method takes advantage of the fact that a transposase can only insert adapter sequences into open chromatin sites. With “deeper” sequencing coverage, it is also possible to distinguish unique binding sites or footprints of transcription factors that may be bound there within the native chromatin.
What’s Next for Epigenetics Research?
With all of the above methods combined with RNA sequencing, it is now possible to directly measure the impact of epigenetic changes using “multi-omic” approaches. For example, combining information of methylation marks, chromatin accessibility, and gene expression may reveal distinct mechanisms driven by the environment in longitudinal studies (analysis of repeat sampling of blood in the same individual as an example)6.
Even more powerful are the applications of these methods together within single cells. With the recent rapid increases in sequencing power paired with greatly decreased costs, interest has spiked in performing single cell epigenomics to generate cell-type specific maps. The Human Cell Atlas project, led by a number of prominent researchers in this cutting edge field, endeavors to create comprehensive reference maps of all human cells. This will allow new levels of understanding of cell development, identity, and function in disease. For example, whole-genome bisulfite sequencing (WGBS) was performed on a single cell of mouse liver and revealed a surprisingly high level of heterogeneity inside liver tissue.7 As sequencing prices continue to drop and new methods are developed, even more exciting epigenetics revelations will soon be on the horizon.
Epigenetic Product Guide
| DNA Methylation Analysis | |
|---|---|
| Bisulfite Methods | |
| Bisulfite conversion | EZ DNA Methylation Kits |
| NGS platforms | |
| PCR platforms | |
| Bisulfite Free Methods | |
| Antibody Methods | |
| Chromatin Analysis | |
| ChIP | |
| Chromatin Structure | Zymo-seq ATAC |
References
- Toraño, E. G.; García, M. G.; Fernández-Morera, J. L.; Niño-García, P.; Fernández, A. F., The Impact of External Factors on the Epigenome: In Utero and over Lifetime. Biomed Res Int 2016, 2016, 2568635.
- Joubert, B. R.; Felix, J. F.; Yousefi, P.; Bakulski, K. M.; Just, A. C.; Breton, C.; Reese, S. E.; Markunas, C. A.; Richmond, R. C.; Xu, C. J.; Küpers, L. K.; Oh, S. S.; Hoyo, C.; Gruzieva, O.; Söderhäll, C.; Salas, L. A.; Baïz, N.; Zhang, H.; Lepeule, J.; Ruiz, C.; Ligthart, S.; Wang, T.; Taylor, J. A.; Duijts, L.; Sharp, G. C.; Jankipersadsing, S. A.; Nilsen, R. M.; Vaez, A.; Fallin, M. D.; Hu, D.; Litonjua, A. A.; Fuemmeler, B. F.; Huen, K.; Kere, J.; Kull, I.; Munthe-Kaas, M. C.; Gehring, U.; Bustamante, M.; Saurel-Coubizolles, M. J.; Quraishi, B. M.; Ren, J.; Tost, J.; Gonzalez, J. R.; Peters, M. J.; Håberg, S. E.; Xu, Z.; van Meurs, J. B.; Gaunt, T. R.; Kerkhof, M.; Corpeleijn, E.; Feinberg, A. P.; Eng, C.; Baccarelli, A. A.; Benjamin Neelon, S. E.; Bradman, A.; Merid, S. K.; Bergström, A.; Herceg, Z.; Hernandez-Vargas, H.; Brunekreef, B.; Pinart, M.; Heude, B.; Ewart, S.; Yao, J.; Lemonnier, N.; Franco, O. H.; Wu, M. C.; Hofman, A.; McArdle, W.; Van der Vlies, P.; Falahi, F.; Gillman, M. W.; Barcellos, L. F.; Kumar, A.; Wickman, M.; Guerra, S.; Charles, M. A.; Holloway, J.; Auffray, C.; Tiemeier, H. W.; Smith, G. D.; Postma, D.; Hivert, M. F.; Eskenazi, B.; Vrijheid, M.; Arshad, H.; Antó, J. M.; Dehghan, A.; Karmaus, W.; Annesi-Maesano, I.; Sunyer, J.; Ghantous, A.; Pershagen, G.; Holland, N.; Murphy, S. K.; DeMeo, D. L.; Burchard, E. G.; Ladd-Acosta, C.; Snieder, H.; Nystad, W.; Koppelman, G. H.; Relton, C. L.; Jaddoe, V. W.; Wilcox, A.; Melén, E.; London, S. J., DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. Am J Hum Genet 2016, 98 (4), 680-96.
- Horvath, S., DNA methylation age of human tissues and cell types. Genome Biol 2013, 14 (10), R115.
- Berdasco, M.; Esteller, M., Clinical epigenetics: seizing opportunities for translation. Nat Rev Genet 2019, 20 (2), 109-127.
- Jones, P. A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D. D., Epigenetic therapy in immune-oncology. Nat Rev Cancer 2019, 19 (3), 151-161.
- Ahadi, S.; Zhou, W.; Schüssler-Fiorenza Rose, S. M.; Sailani, M. R.; Contrepois, K.; Avina, M.; Ashland, M.; Brunet, A.; Snyder, M., Personal aging markers and ageotypes revealed by deep longitudinal profiling. Nat Med 2020, 26 (1), 83-90.
- Gravina, S.; Dong, X.; Yu, B.; Vijg, J., Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome. Genome Biol 2016, 17 (1), 150.