A Quick Guide for DNA Methylation Profiling with NGS-based Methods
What is DNA Methylation – An Epigenetic Modification that Regulates Gene Expression
Did you know that the amount of cuddles a baby receives from a caregiver could affect the baby’s DNA methylation profile, and in turn, the developmental progress?1 In fact, DNA methylation affects various aspects of daily life that most people may not even think about, such as the ripening process of sweet orange fruits.2 So, what is DNA methylation?
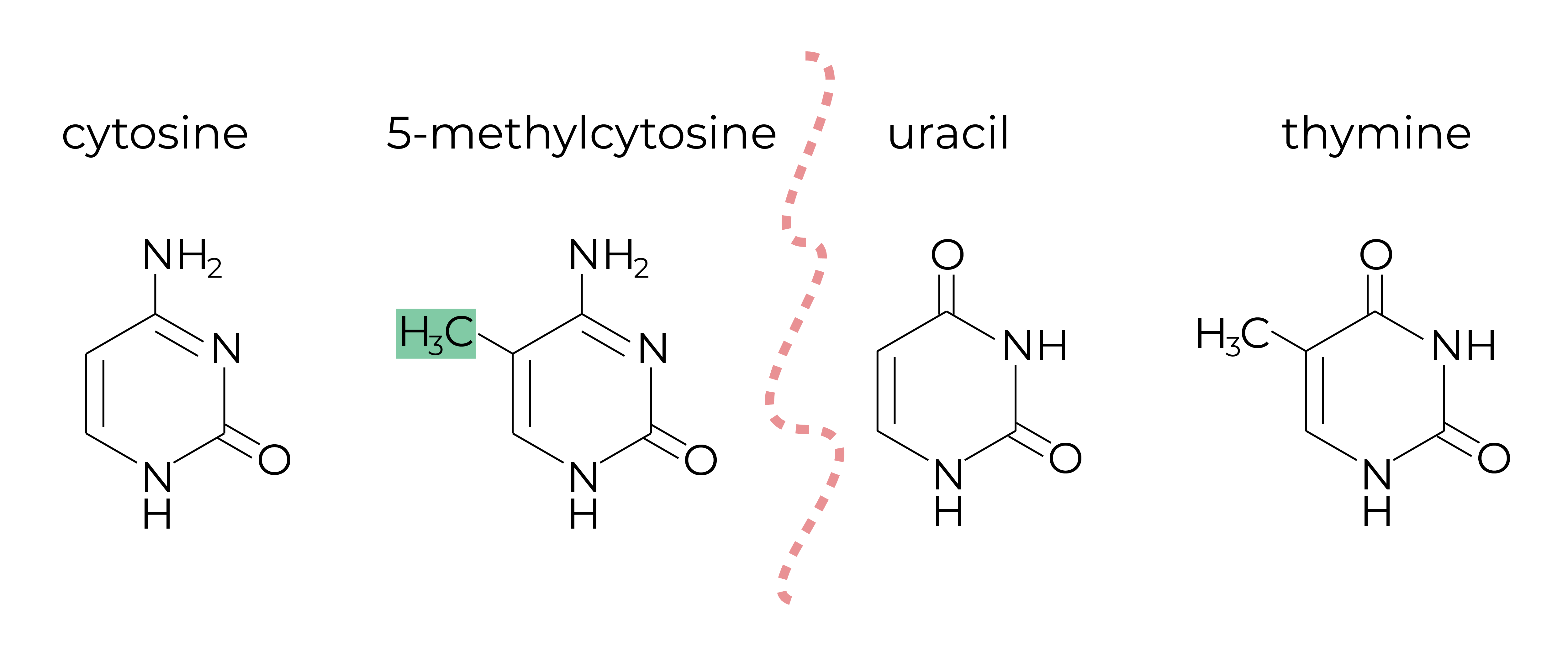
DNA methylation is a chemical modification to DNA molecules. In most cases, DNA methylation refers to the covalent addition of a methyl group to the 5-carbon position of cytosine, forming 5-methylcytosine (5mC) (Figure 1, the two structures on the left). DNA methyltransferases perform this reaction to establish and maintain DNA methylation patterns across the genome. DNA methylation regulates gene expression without changing the sequences of the nucleotides. Hence, DNA methylation is an epigenetic mark, where “epi” implies that this feature is “on top of” genetics playing its roles without alterations in the DNA sequence.
DNA methylation is among the most extensively studied epigenetic marks.3 In eukaryotes, DNA methylation pervasively occurs at CpG dinucleotides. The patterns of DNA methylation at promoters, gene bodies and enhancers play vital roles in regulating gene expression.4-6
Commonly Recognized DNA Methylation Patterns
Some DNA methylation patterns have been widely studied and researchers have acquired a fair amount of knowledge on their regulation of gene expression. A few examples of such DNA methylation patterns are as follows:
- DNA hypermethylation (substantially increased DNA methylation levels) in promoter regions is generally associated with gene suppression or silencing. A good example is that DNA hypermethylation serves to inactivate the X chromosome and silence repetitive DNA sites.4
- The CpG sites in CpG islands (CGIs, regions with high frequency of GpG sites) associated with the promoters of housekeeping genes and developmental regulatory genes are usually resistant to DNA methylation. However, these promoter CGIs were found to be aberrantly hypermethylated in tumor suppressor genes and DNA mismatch repair genes, which in turn suppress the related genes’ activation and expression. Such aberrant DNA methylation pattern in promoter CGIs is perhaps the most widely studied epigenetic alteration in cancers.5, 6
- CpGs in enhancers have been found to contain tissue- or cell type-specific DNA methylation patterns. Such differentially methylated regions (DMRs) may have contributed to the unique gene expression among different tissue and cell types.7
Given that precise gene regulation is important for many fundamental biological processes, DNA methylation is essential throughout organismal development and homeostasis. Therefore, elucidating DNA methylation patterns in samples from various species under diverse conditions enables further exploration of its roles in gene regulatory mechanisms. This will moreover enhance our understanding in how DNA methylation influences human diseases and aging, as well as the health of ecosystems. How then, do we profile DNA methylation?
How to Profile DNA Methylation
The gold-standard reaction for DNA methylation analysis is called bisulfite conversion. Bisulfite reagents convert unmethylated cytosine residues to uracils (Figure 1, the third structure) while leaving the methylated ones untouched. During PCR, DNA polymerase recognize uracils as thymines and the subsequent analysis takes “C” as methylated cytosines while “T” (Figure 1, the last structure) as unmethylated cytosines in the DNA sample. Many methods have been developed to investigate DNA methylation-based on bisulfite conversion. Since the demand of genome-wide DNA methylation profiling has increased, NGS-based methods have become much more popular.
Methods Based-on Next-Generation Sequencing (NGS). NGS-based methods are the most comprehensive and have emerged as the primary means of DNA methylation analysis recently. NGS-based methods produce results with high coverages across an entire genome and are compatible with any species. Bisulfite sequencing is thus gaining tremendous popularity among researchers for DNA methylation profiling, indicated by the increasing number of related publications over the years. DNA samples that are bisulfite converted must be constructed into libraries that contain adapters for sequencing on a compatible NGS instrument. The methylated cytosines are recognized as “C” in NGS readouts.
Thus far, researchers have implemented a myriad of bisulfite sequencing methods to uncover DNA methylation information for diverse biological questions. The most extensively used NGS methods include reduced representation bisulfite sequencing (RRBS), whole-genome bisulfite sequencing (WGBS), and targeted bisulfite sequencing. These methods share common steps in the procedure of library preparation such as bisulfite conversion, adapter ligation and PCR amplification, but differ slightly at the beginning of the workflows preparing the input DNA materials.
Reduced Representation Bisulfite Sequencing (RRBS)
- RRBS uses restriction enzyme digestion to enrich for CpG-rich regions across the genome from the input DNA before proceeding to the common steps in bisulfite sequencing library preparation.
- Features of RRBS.
- RRBS reduces the number of required sequencing reads to achieve similar data quality compared to sequencing the entire bisulfite converted genome as in WGBS, and reveals DNA methylation information on most important regulatory regions such as promoters, CpG islands, and in gene bodies. With approximately 10-20% of the sequencing reads that are normally required by WGBS, RRBS can cover ≥70% of promoters, CpG islands, gene bodies, and around 35% of enhancers.
- RRBS is more cost-effective compared to WGBS in sequencing and is ideal for scientists to screen genome-wide DNA methylation information in large-scale studies.
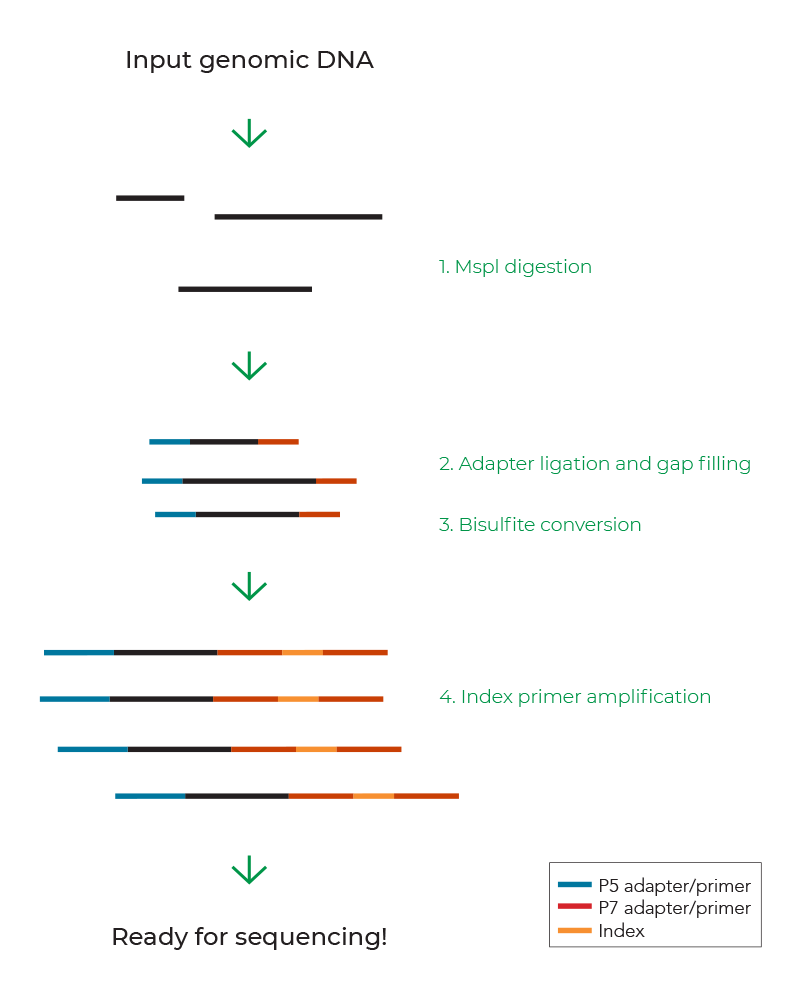
- Many variations of RRBS have emerged over the years and an example of the basic RRBS workflow in Figure 2 covers the main steps to prepare RRBS libraries from input genomic DNA: MspI digestion, adapter ligation, bisulfite conversion, and PCR amplification with indexed primers. This specific kit has a simple protocol and is compatible with as low as 10 ng of genomic DNA, making RRBS more accessible to all users.8-11
Whole-Genome Bisulfite Sequencing (WGBS) Expands The Frontiers Of Genomic Medicine
WGBS detects the methylation status of nearly all the cytosines in the input sample. As a result, it offers scientists a comprehensive profile of DNA methylation levels across the entire genome. The utilization of WGBS data enhances the likelihood of unveiling novel DNA methylation patterns through de novo discovery.12,13
Depending on the genome's size, WGBS might necessitate several hundreds of millions of sequencing reads to achieve sufficient depth for accurate methylation analysis. The advent of new sequencing technologies has substantially lowered the cost associated with WGBS.
The increased cost-effectiveness of WGBS positions it as the primary discovery platform for identifying markers in targeted clinical applications.

Whole-Genome Bisulfite Sequencing (WGBS) Preparation In One Tube
Various workflows are accessible for WGBS (Whole-Genome Bisulfite Sequencing). While their specifics may vary, these workflows typically encompass optional fragmentation of input genomic DNA, bisulfite DNA treatment, adapter ligation, and PCR amplification with indexed primers.
The conventional WGBS workflow is insufficient to meet the growing demand for robustness in library preparation.
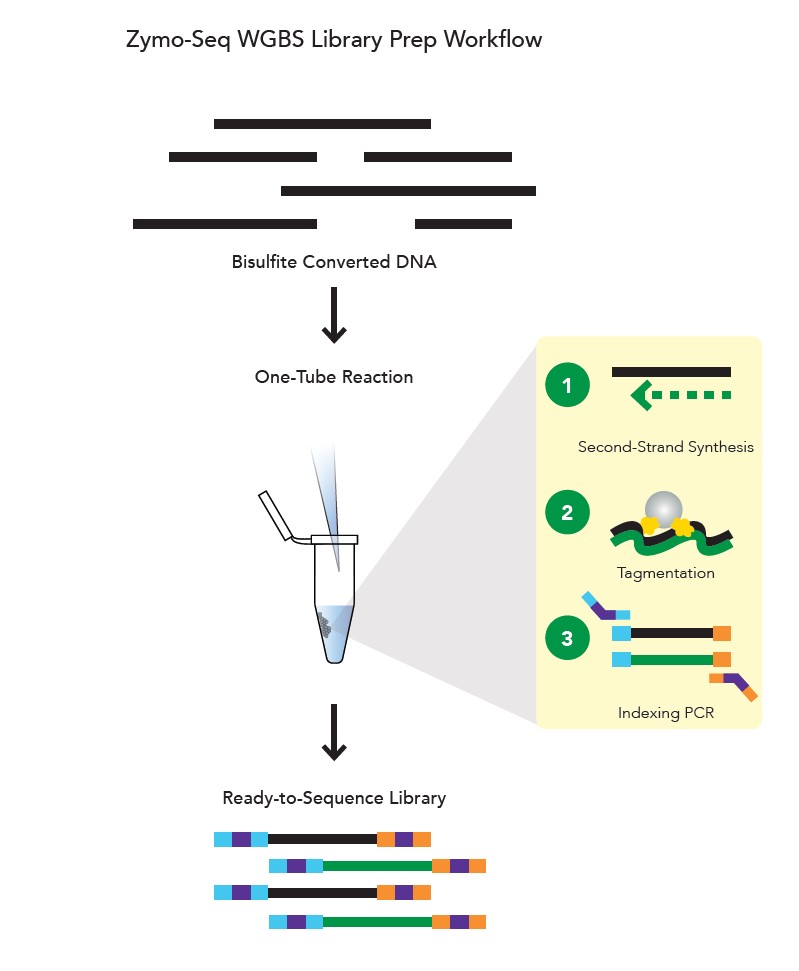
The Zymo-Seq WGBS Library Kit addresses the escalating requirements for sample throughput and minimizes sample loss. It stands as the sole kit designed for whole-genome bisulfite (WGBS) library preparation within a single tube. The Zymo-Seq WGBS Library Kit integrates tagmentation technology to eliminate the laborious fragmentation, enzymatic, and clean-up steps inherent in traditional ligation-based library preparation methods. This streamlined workflow reduces hands-on time, rendering it an optimal choice for applications with higher throughput demands.14
General WGBS workflow is shown in Figure 4.
High Quality WGBS Libraries from Precious Clinical Samples
Since the initial discovery of circulating cell-free DNA (cfDNA), numerous studies have illuminated a compelling correlation between DNA methylation biomarkers in cfDNA samples and tumor presence. Notably, cancer patients frequently exhibit significantly distinct DNA methylation patterns in their cfDNA, particularly tumor-derived cfDNA (ctDNA), when compared to individuals without cancer. This revelation has ignited a surge of interest in harnessing DNA methylation analysis of cfDNA for applications such as cancer detection, monitoring disease progression, selecting appropriate treatments, and tracking treatment efficacy.
Due to the inherent limitations of sample size in liquid biopsies, next-generation sequencing-based molecular profiling has emerged as a pivotal approach for detecting cancer-associated DNA methylation alterations in cfDNA. This method enables the identification of crucial epigenetic changes linked to cancer development and progression.
Beyond cancer, ongoing research is also exploring the application of DNA methylation biomarkers in cfDNA for diverse purposes, including prenatal genetic testing, monitoring organ transplant outcomes, and assessing recovery following conditions like burns or stroke.
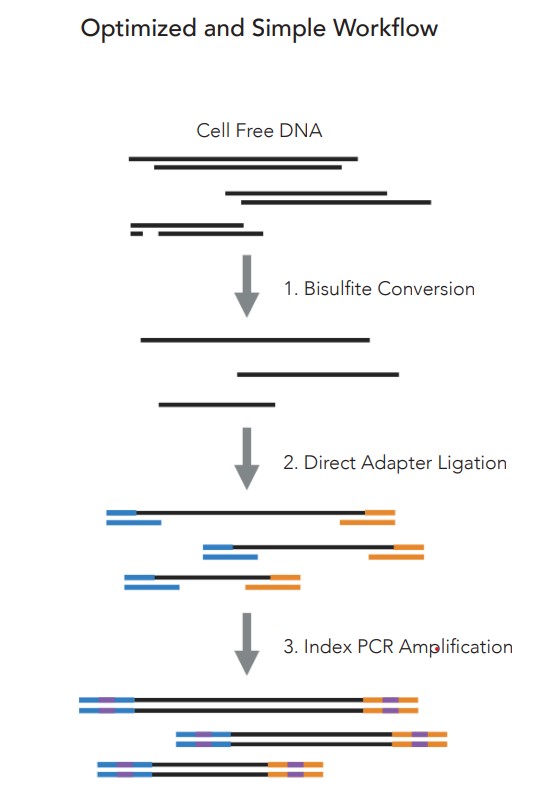
The Zymo-Seq Cell Free DNA WGBS Library Kit is optimized to create high quality WGBS libraries from as little as 5 ng of cfDNA. This innovative NGS kit directly captures and ligates the adapters onto any size DNA fragment, facilitating sequencing of nicked, damaged, and short cfDNA fragments. The direct ligation also eliminates the need for end repair and dA tailing steps, thus reducing bias by preserving the integrity of any native methylation that is present on the fragment termini.
Zymo-seq Cell free WGBS workflow is shown in Figure 5.
Pushing the Limit of Ultra Low Input WGBS Library Prep
In some cases, researchers may have access to only a very small amount of DNA, such as rare cell populations, clinical samples, or historical specimens. Ultra-low input WGBS allows them to study the DNA methylation patterns in these limited samples.
Single-cell epigenomics is a growing field that focuses on understanding DNA methylation patterns at the single-cell level. Ultra-low input WGBS is essential for studying DNA methylation in individual cells, uncovering cell-to-cell variability, and understanding cellular heterogeneity.
In summary, ultra-low input WGBS is crucial for researchers across various fields, such as cancer researchers, developmental biologists, neuroscientists, historical and archaeological scientists who work with limited or precious samples.
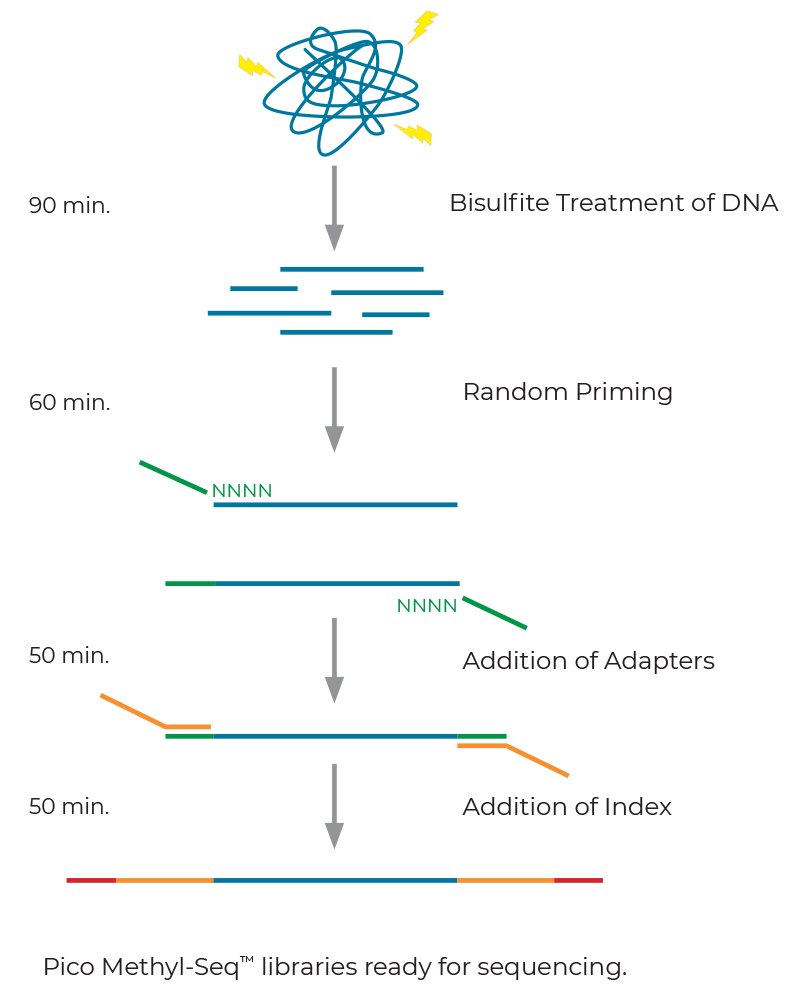
Pico Methyl-Seq Library Prep Kit is particularly outstanding in its compatibility with small amount of gDNA input as low as 10 pg. And it is suitable for single-cell applications where DNA quantity is a limiting factor.15-17
Pico methyl-seq library prep kit workflow is shown in Figure 6.
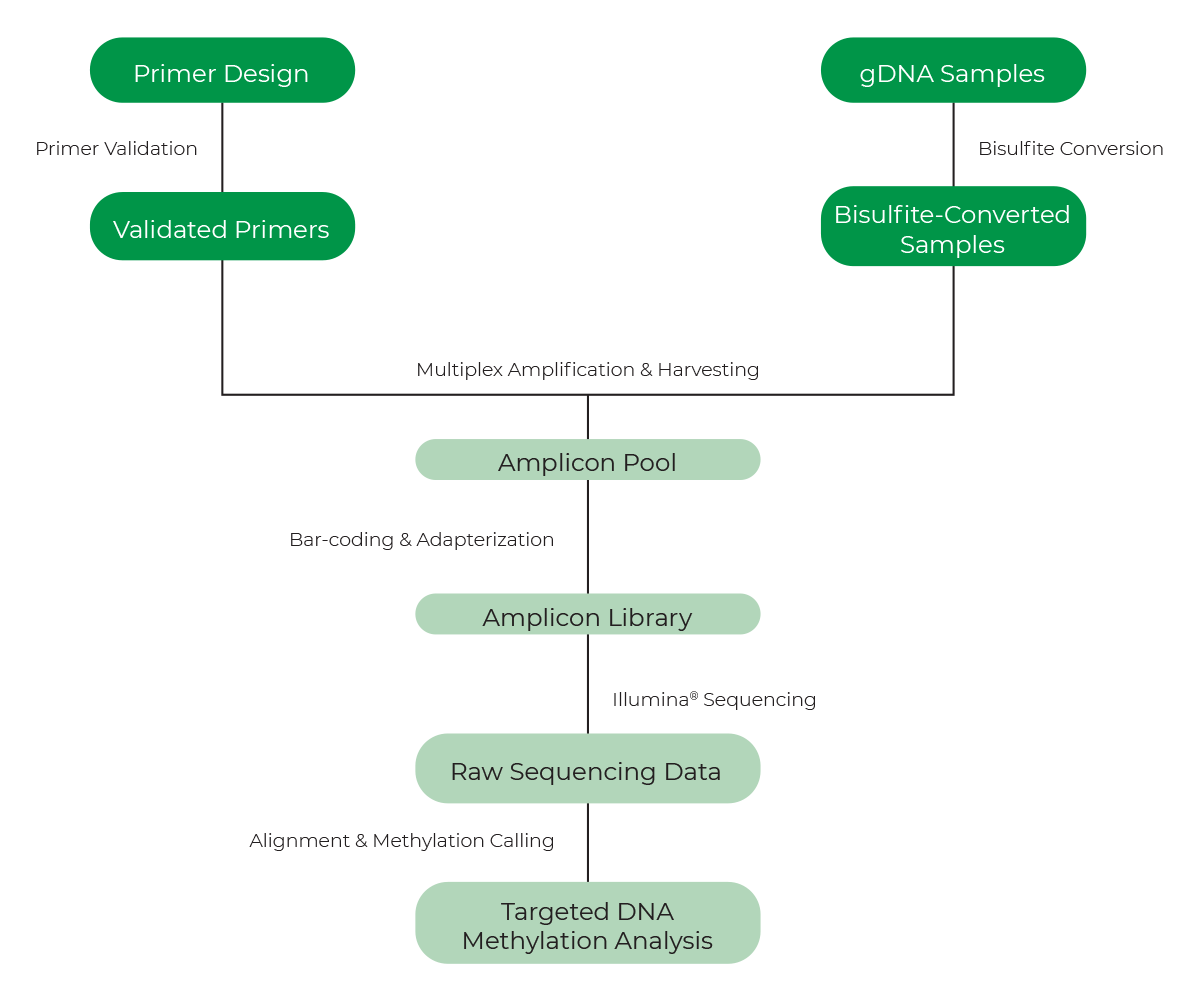
Targeted Bisulfite Sequencing
- Besides the two previously mentioned methods for genome-wide DNA methylation profiling, targeted bisulfite sequencing enables site-specific DNA methylation detection in desired loci.
- Features of targeted bisulfite sequencing:
- The most direct way is to design NGS-compatible PCR primers for specific gene regions of interest to generate libraries. This allows the investigation of DNA methylation levels in only these selected genomic regions, therefore, reducing per sample sequencing costs as compared to genome-wide based strategies.
- Targeted bisulfite sequencing is a great option for validation of data generated by other genome-wide methods as well as screening on multi-targeted gene regions in large sample cohorts. Scientists can examine from 1 to 192 regions and beyond in the same sample using this method.
- A general workflow of the targeted bisulfite sequencing available from Zymo Research is shown in Figure 7.
See below for a selection of Zymo Research products and services for DNA methylation study. We have a comprehensive solution for DNA methylation profiling, for every application and workflow.
| Applications | Recommended Kit or Service |
|---|---|
| Reduced Representation Bisulfite Sequencing (RRBS) | Zymo-Seq RRBS Library Kit |
| Whole-genome bisulfite sequencing (WGBS) | |
| Targeted Bisulfite Sequencing | MethylCheck™ DNA Methylation Validation Platform |
| Bisulfite Conversion | EZ DNA Methylation-Lightning Kit (free sample available) |
| Bisulfite Conversion directly from cells | EZ DNA Methylation-Direct Kits |
| DNA Extraction | |
| DNA Clean up | |
| DNA Standards for Methylation Study |
References
- Moore, S. R.; McEwen, L. M.; Quirt, J.; Morin, A.; Mah, S. M.; Barr, R. G.; Boyce, W. T.; Kobor, M. S., Epigenetic correlates of neonatal contact in humans. Dev Psychopathol 2017, 29 (5), 1517-1538.
- Huang, H.; Liu, R.; Niu, Q.; Tang, K.; Zhang, B.; Zhang, H.; Chen, K.; Zhu, J. K.; Lang, Z., Global increase in DNA methylation during orange fruit development and ripening. Proc Natl Acad Sci U S A 2019, 116 (4), 1430-1436.
- Smith, Z. D.; Meissner, A., DNA methylation: roles in mammalian development. Nat Rev Genet 2013, 14 (3), 204-20.
- Greenberg, M. V. C.; Bourc'his, D., The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol 2019, 20 (10), 590-607.
- Flavahan, W. A.; Gaskell, E.; Bernstein, B. E., Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357 (6348).
- Ehrlich, M., DNA hypermethylation in disease: mechanisms and clinical relevance. Epigenetics 2019, 14 (12), 1141-1163.
- Song, Y.; van den Berg, P. R.; Markoulaki, S.; Soldner, F.; Dall'Agnese, A.; Henninger, J. E.; Drotar, J.; Rosenau, N.; Cohen, M. A.; Young, R. A.; Semrau, S.; Stelzer, Y.; Jaenisch, R., Dynamic Enhancer DNA Methylation as Basis for Transcriptional and Cellular Heterogeneity of ESCs. Mol Cell 2019, 75 (5), 905-920.e6.
- Zhao, B.; van Bodegom, P. M.; Trimbos, K. B. Environmental DNA methylation of Lymnaea stagnalis varies with age and is hypermethylated compared to tissue DNA. Mol Ecol Resour 2023, 23 (1), 81-91.
- Venkatesh, G.; Tönges, S.; Hanna, K.; Ng, Y. L.; Whelan, R.; Andriantsoa, R.; Lingenberg, A.; Roy, S.; Nagarajan, S.; Fong, S.; et al. Context-dependent DNA methylation signatures in animal livestock. Environ Epigenet 2023, 9 (1), dvad001.
- Gurao, A.; Vasisth, R.; Singh, R.; Dige, M. S.; Vohra, V.; Mukesh, M.; Kumar, S.; Kataria, R. S. Identification of differential methylome signatures of white pigmented skin patches in Nili Ravi buffalo of India. Environ Mol Mutagen 2022, 63 (8-9), 408-417.
- Ben-Nun, O.; Kisliouk, T.; Marco, A.; Rosenberg, T.; Meiri, N. Early-life thermal stress mediates long-term alterations in hypothalamic microglia. Glia 2022, 70 (4), 619-633.
- Zhou, L.; Ng, H. K.; Drautz-Moses, D. I.; Schuster, S. C.; Beck, S.; Kim, C.; Chambers, J. C.; Loh, M. Systematic evaluation of library preparation methods and sequencing platforms for high-throughput whole genome bisulfite sequencing. Sci Rep 2019, 9 (1), 10383.
- Sun, Z.; Cunningham, J.; Slager, S.; Kocher, J. P. Base resolution methylome profiling: considerations in platform selection, data preprocessing and analysis. Epigenomics 2015, 7 (5), 813-828.
- Venkataraman, Y. R.; White, S. J.; Roberts, S. B. Differential DNA methylation in Pacific oyster reproductive tissue in response to ocean acidification. BMC Genomics 2022, 23 (1), 556.
- Nurislamov, A.; Lagunov, T.; Gridina, M.; Krasikova, A.; Fishman, V. Single-Cell DNA Methylation Analysis of Chicken Lampbrush Chromosomes. Int J Mol Sci 2022, 23 (20).
- Michki, N. S.; Ndeh, R.; Helmin, K. A.; Singer, B. D.; McGrath-Morrow, S. A. DNA methyltransferase inhibition induces dynamic gene expression changes in lung CD4. Sci Rep 2023, 13 (1), 4283.
- Fang, S.; Cui, D.; Hong, T.; Guo, L.; Lee, Y. T.; Lee, M.; Isgandarova, S.; Martinez-Moczygemba, M.; Zhou, Y.; Li, J.; et al. Ten-Eleven Translocation Ablation Impairs Cardiac Differentiation of Mouse Embryonic Stem Cells. Stem Cells 2022, 40 (3), 260-272.