Zymo Research Takes Aim at Shotgun Metagenomic Sequencing
The ever-increasing depth achieved using Next-Gen sequencing in the field of microbiomics is providing more information about how microbes affect our lives and environment. As sequencing costs continue to drop, microbiome characterization with shotgun metagenomic sequencing is gaining increased attention.
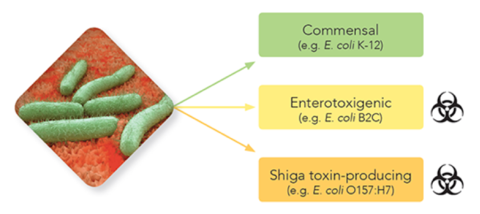
Compared with targeted sequencing (e.g. 16S, 18S or ITS sequencing), advantages of shotgun metagenomic sequencing include strain-level resolution, across-kingdom identification, and functional gene profiling. While useful for all microbiome studies, strain-level resolution is critical for the identification of pathogenic microbes apart from benign commensals of the same species.
Further, pathogen identification across kingdoms including eubacteria, fungi, and protists is key to understanding microbiome community profiling for diagnosis of infectious disease. Furthermore, gene function profiling, rather than taxonomy alone, is important for the characterization of microbial communities. For example, identification of virulence factors and antibiotic resistance genes provides valuable information for the treatment of infectious diseases.
Despite the advantages, shotgun sequencing is not without limitations. Similar to other NGS-based measurements, the workflow has multiple steps from sample collection to bioinformatics analysis. Without proper consideration, bias and errors are easily introduced at each step which will compound and skew the analysis.
Without proper sample protection and storage, microbiome composition can shift away from that at the time of collection. Recently, the American Gut Project observed bacterial blooms during fecal sample transportation and the researchers were forced to address the impact with bioinformatics [1]. This approach fundamentally skews the analysis by potentially eliminating genomic information which would not be removed if the sample is properly preserved at the time of collection.
Also, DNA extraction methods can introduce dramatic bias. In fact, recently it was demonstrated that the protocols used by the Human Microbiome Project (HMP) and the Metagenomics of the Human Intestinal Tract (MetaHIT) caused different profiling results of the same fecal samples, especially regarding the abundance of Bacteroidetes [2]. The root cause of this difference was most likely due to methodological biases causing differences in the proportional abundances of easy-to-lyse and tough-to-lyse microbes. Therefore, without challenge to the methodologies used in each study to ensure proper lysis of all microbes, these studies become incomparable.
When performed properly, shotgun metagenomic sequencing generally has less bias than 16S sequencing. However, all library preparation processes contain procedures which can lead to bias. For example, sequence coverage can vary between species due to differences in genomic GC content. Additionally, one major challenge in shotgun metagenomics resides in the bioinformatics analysis. For example, McIntyre and colleagues compared 11 different pipelines for shotgun metagenomics analysis and found dramatic inconsistency in their performance [3]. The performance of these pipelines in terms of the rates of false positives and false negatives varies greatly. Besides the algorithm used for taxonomy prediction, the comprehensiveness of the reference database plays an important role in accurately identifying the source of sequencing reads.
Although the strain-level, cross-kingdom identification, and gene profiling capabilities of shotgun metagenomic sequencing offer advanced solutions for disease diagnosis and microbiome profiling, biases introduced at each step of the workflow can distort resorts. To provide the highest quality shotgun metagenomics analysis to the scientific community, Zymo Research provides reagents for unbiased and accurate sample collection, DNA extraction, library preparation, and sequencing. This portfolio is rigorously tested and validated using the ZymoBIOMICS® microbiome standards.
Furthermore, the ZymoBIOMICS services then empowers accurate bioinformatics analysis with sophisticated algorithms and the world’s largest curated microbiome database (> 150,000 genomes). This results in strain-level identification, across-kingdom (Bacteria, fungi, virus, and parasites) identification, and functional gene profiling (e.g. antibiotics resistance genes and virulence genes).
The Zymo Research metagenomics solution offers the simplest, most powerful collection-to-conclusion solution available.
Click here to inquire about ZymoBIOMICS® shotgun metagenomic sequencing services
References:
1. Amir A, McDonald D, Navas-Molina JA, Debelius J, Morton JT, Hyde E, et al. Correcting for Microbial Blooms in Fecal Samples during Room-Temperature Shipping. mSystems. 2017;2.
2. Wesolowska-Andersen A, Bahl MI, Carvalho V, Kristiansen K, Sicheritz-Ponten T, Gupta R, et al. Choice of bacterial DNA extraction method from fecal material influences community structure as evaluated by metagenomic analysis. Microbiome. 2014;2:19.
3. McIntyre ABR, Ounit R, Afshinnekoo E, Prill RJ, Henaff E, Alexander N, et al. Comprehensive benchmarking and ensemble approaches for metagenomic classifiers. Genome Biol. 2017;18:182.





