
What is ATAC-Seq?
ATAC-Seq (Assay for Transposase-Accessible Chromatin using sequencing) is a high-throughput epigenomic technique used to analyze chromatin accessibility, offering insights into gene regulation, transcription factor binding, and epigenetic mechanisms.
This method uses a hyperactive Tn5 transposase enzyme to target and fragment open chromatin regions while simultaneously inserting sequencing adapters in a one-step process called tagmentation. These regions are typically enriched for active regulatory elements, such as enhancers and promoters (Buenrostro et al., 2015).
The streamlined workflow of ATAC-Seq—especially its minimal input requirements—has made it a widely adopted method in functional genomics, single-cell epigenomics, and disease research.
ATAC-Seq is also widely regarded as both robust and cost-effective. The simplicity of the protocol—requiring fewer enzymatic steps, minimal starting material, and no need for complex antibody pulldowns (as in ChIP-Seq)—reduces technical variability and hands-on time. These features make it well-suited for labs with limited budgets, high-throughput project demands, or those working with rare or precious samples. Moreover, ATAC-Seq provides high-resolution data with excellent signal-to-noise ratio, making it a reliable choice for routine chromatin accessibility profiling across diverse cell types and organisms.
Overview of the ATAC-Seq Library Prep Workflow
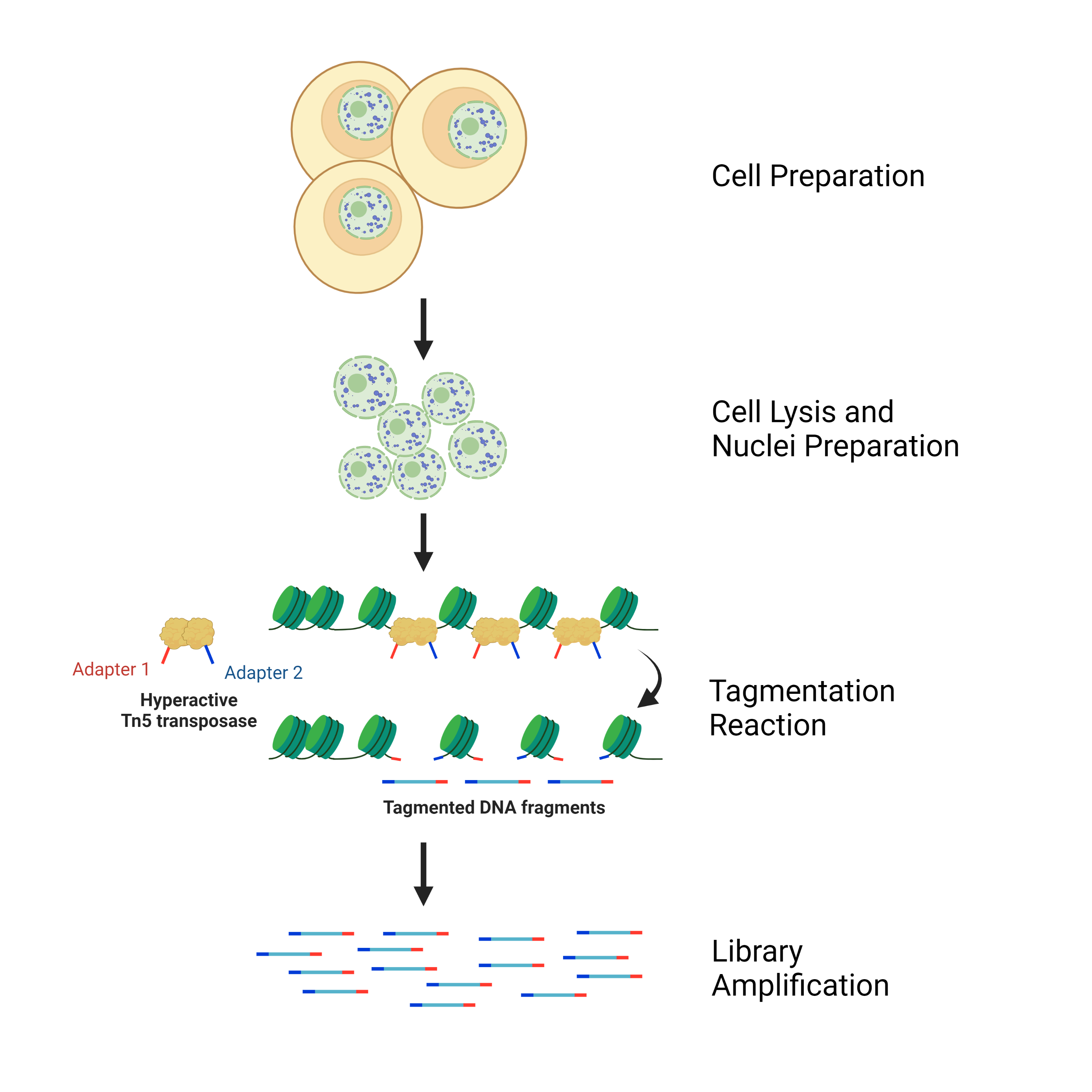
The ATAC-Seq workflow traditionally begins with the preparation of live cultured cells. The chromatin within cells is naturally structured into a tightly packed arrangement of nucleosomes with regulatory regions exposed in open sections of the chromatin. To capture and sequence these accessible regions, cells are lysed with a gentle detergent to release their nuclei while still maintaining the overall structure of the chromatin. Isolated nuclei are then added to a tagmentation reaction that utilizes Tn5 transposase to fragment open chromatin regions while simultaneously tagging these fragments with a sequencing adapter. This reaction is then purified before proceeding to an index PCR step where the fragments are amplified as well as indexed with a sequencing barcode. After one final clean up, one is left with an ATAC-Seq library ready to be sequenced.
Difficult Sample Types
While ATAC-Seq is a powerful tool for studying chromatin accessibility, certain sample types can present unique challenges that require special adaptations to prepare quality libraries. Nontraditional and difficult sample inputs suffer from issues ranging from low cell count, DNA degradation, difficult nuclei isolation steps, contamination, and more. Researchers have developed various strategies to overcome these obstacles, enabling ATAC-Seq to be applied to a wide variety of biological samples.
Animal tissues are among the most common and challenging sample types for ATAC-Seq library preparation, due to their diverse structures and biochemical complexities. In particular, neurons and brain tissue present significant difficulties stemming from their intricate nuclear architecture, densely packed chromatin, and elevated lipid content. The high lipid concentration can obstruct efficient nuclei isolation and hinder the tagmentation reaction, resulting in reduced chromatin accessibility signal and increased background noise (Kim, 2023). To improve data quality in these samples, researchers frequently optimize the protocol by increasing Tn5 transposase enzyme concentration, extending tagmentation incubation times, and modifying lysis buffer composition to more effectively solubilize lipid-rich membranes.
Muscle tissue presents a distinct set of challenges for ATAC-Seq due to its dense and fibrous composition. Rich in connective tissue components such as collagen and fibronectin, muscle samples are often tough and resistant to dissociation, complicating the process of nuclei isolation. To generate high-quality chromatin accessibility data from these tissues, researchers typically employ mechanical disruption techniques, including gentle douncing or homogenization, in combination with detergent-based lysis buffers. These steps help break down the extracellular matrix and facilitate the release of intact nuclei, ensuring a more efficient and reproducible ATAC-Seq workflow for fibrous tissue samples.
Plant tissues pose unique technical hurdles for ATAC-Seq, largely due to their rigid cell walls and biochemical complexity. The presence of secondary metabolites and high levels of organellar DNA, particularly from chloroplasts and mitochondria, can significantly compromise data quality. One major issue is that the hyperactive Tn5 transposase used in the tagmentation step does not discriminate between nuclear and extranuclear DNA, leading to the unintended fragmentation and sequencing of non-nuclear genomes. This reduces the proportion of informative reads that align to the nuclear genome and diminishes the accuracy of open chromatin region identification (Bajic, 2018).
To address these challenges, researchers focus heavily on optimizing the nuclei isolation protocol. Two commonly used methods include sucrose sedimentation, which separates nuclei from debris via centrifugation through density gradients, and the INTACT (Isolation of Nuclei Tagged in specific Cell Types) technique, which uses streptavidin-coated magnetic beads to isolate biotinylated nuclei from complex cell lysates (Deal, 2011). Both strategies are designed to enrich clean, intact nuclei and minimize contamination from organelle-derived DNA, ultimately improving the efficiency and reliability of ATAC-Seq in plant samples.
Conclusion
ATAC-Seq continues to be a transformative method for profiling chromatin accessibility, enabling researchers to gain valuable insights into gene regulation across a wide range of organisms and sample types. With thoughtful optimizations, even the most difficult tissues and cell types can yield high-quality data. The Zymo-Seq ATAC-Seq Library Kit offers an effective and accessible solution for scientists working with low-input or complex samples. To learn more or explore product specifications, visit the Zymo-Seq ATAC-Seq Library Kit page.
Citations
- Bajic, M., Maher, K. A., & Deal, R. B. (2018). Identification of open chromatin regions in plant genomes using ATAC-seq. In J. T. Loveys (Ed.), Methods in molecular biology (Vol. 1675, pp. 183–201). Humana Press. https://doi.org/10.1007/978-1-4939-7318-7_12
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., & Greenleaf, W. J. (2015). ATAC-seq: A method for assaying chromatin accessibility genome-wide. Current protocols in molecular biology, 109, 21.29.1–21.29.9. https://doi.org/10.1002/0471142727.mb2129s109
- Deal, R. B., & Henikoff, S. (2011). The INTACT method for cell type-specific gene expression and chromatin profiling in Arabidopsis thaliana. Nature Protocols, 6(1), 56–68. https://doi.org/10.1038/nprot.2010.175
- Kim, K., Choi, Y., Lee, S., Kim, J., Kim, H., Cho, S., & Park, K. (2023). Adipocyte-specific ATAC-seq with adipose tissues using fluorescence-activated nucleus sorting. Journal of Visualized Experiments, 193, e65033. https://doi.org/10.3791/65033